
We heavily rely on application programming interfaces (APIs) for searching, performing tasks, and exchanging data more than ever before. This is no different in the healthcare domain, which is becoming even more important with the advent of data exchange protocols such as the Beacon protocol from the Global Alliance for Genomics and Health (GA4GH). In this article, we will briefly discuss how we can apply burgeoning GenAI products, especially large language models (LLMs), to make health APIs more accessible. We will use the Beacon API as the basis.
Current approach and the opportunity
Typically, an API like the Beacon API is accessed using an REST API client such as Postman or using a dedicated web interface such as the PathSBeacon interface of any web interface. For this article we will use a mock user interface that is capable of performing Beacon related phenotypic and genotypic queries. Consider the following two question.
What are the variants in the 1st chromosome from base pairs 500 kb to 501 kb?
Who are the individuals with renal failure and liver damage where the samples were isolated using a broth medium?
These two questions essential target two different scopes; (1) genomic variants and (2) individual information.
The first questions may be submitted to an API using an interface or a JSON (javascript object notation) as follows.
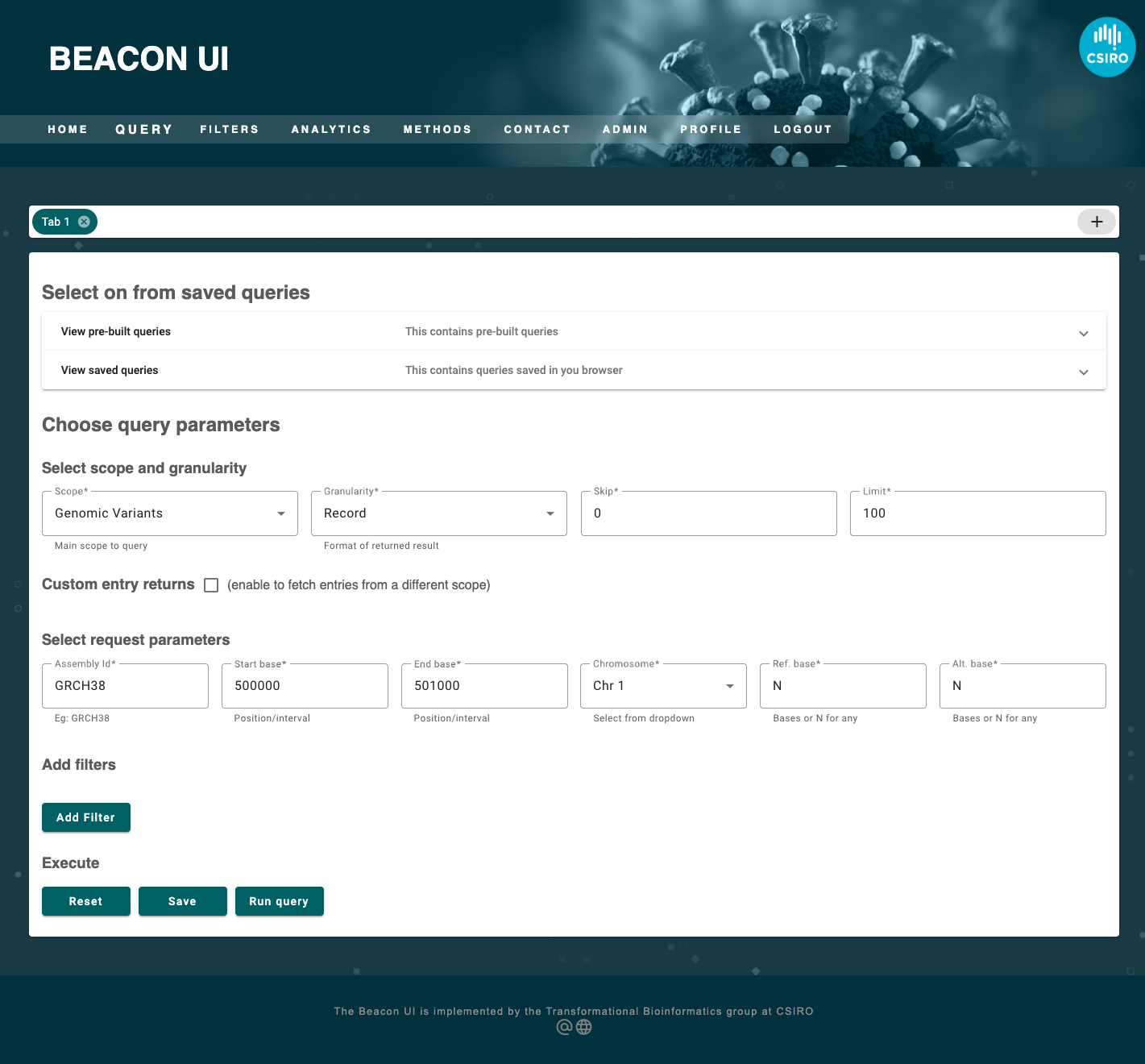
{
"query": {
"requestParameters": {
"assemblyId": "GRCH38",
"start": [
"500000"
],
"end": [
"501000"
],
"referenceName": "1",
"referenceBases": "N",
"alternateBases": "N"
}
}
...
}
POST to /g_variants
For the second query it would be as follows.
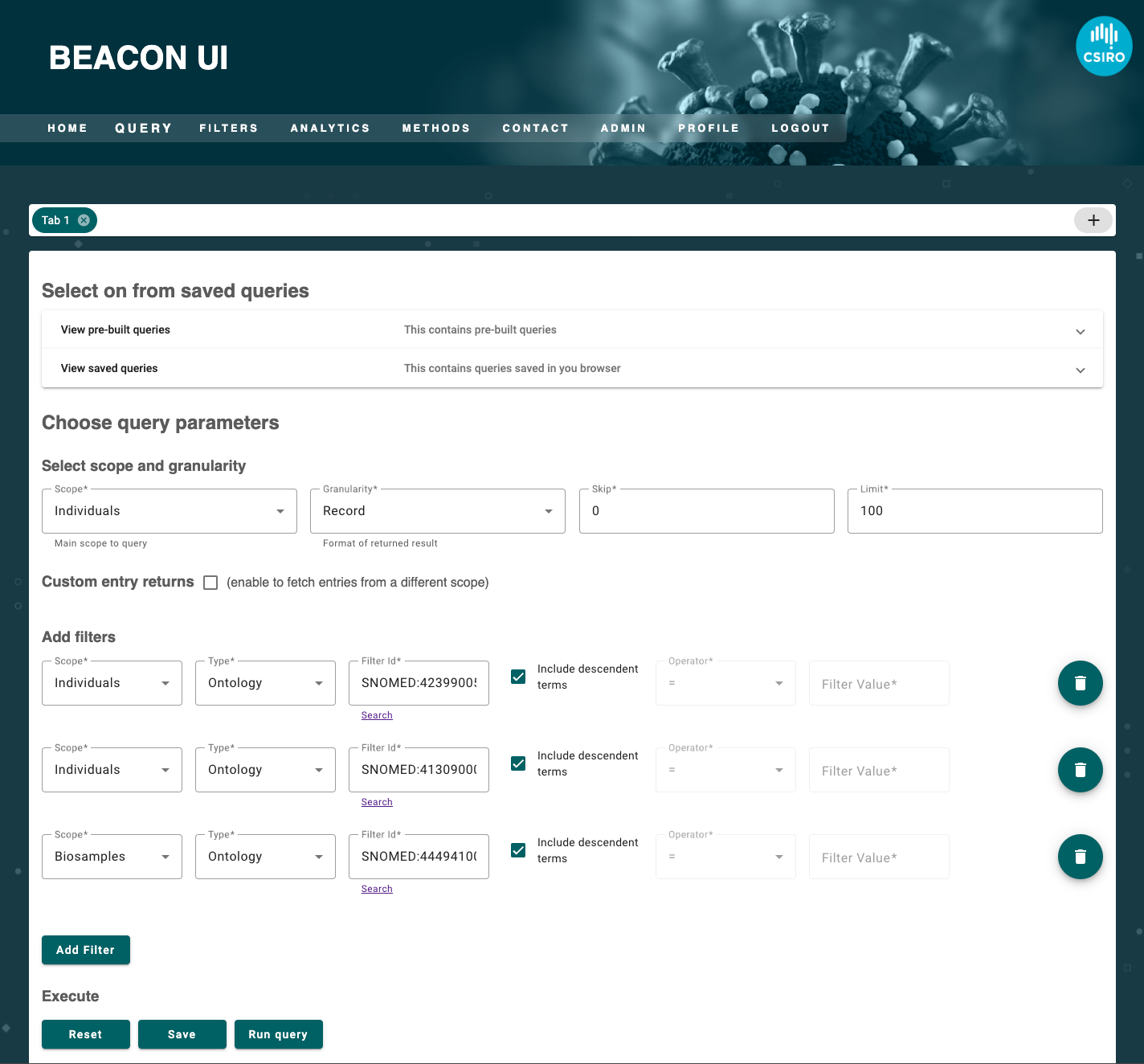
{
"query": {
"filters": [
{
"scope": "individuals",
"id": "SNOMED:42399005"
},
{
"scope": "individuals",
"id": "SNOMED:41309000"
},
{
"scope": "biosamples",
"id": "SNOMED:444941003"
}
]
...
}
...
}
POST to /individuals
In both scenarios, users must either navigate the user interface to fill the form fields or generate a JSON object on their own. However, JSON is the only form that API understands and natural language is the main medium all of us use to communicate. At the same time, visually filled forms is the best way for us to read the query in a well structured manner. The opportunity further broadens as multiple coding mechanism get involved in a beacon, for example interpreting renal failure using SNOMED (SNOMED:42399005) or NCIT (NCIT:C4376) or HPO (HP:0004713 - reversible renal failure) in mix for storing phenotypic information.
Bridging the gap using LLMs
During the recent years LLMs which is a class of GenAI (generative artificial intelligence) models showed us promising capabilities in understanding the natural language in assisting day-to-day tasks. Furthermore, LLMs are known to be quite good at code generation, which certainly makes them quite good with generating JSON objects.
The anatomy of the beacon query
From the above examples it is clearly evident that those two different queries must interrogate two different endpoints based on the scope of the query. Consequently, if the query were to seek variants it should optionally extract the parameters applicable to a variant query such as start, end, chromosome and assembly_id (grch37, grch38, etc) associated. For both kinds of queries we must extract filters in the form of objects having a coded term and a scope. For simplicity, we will only consider filters for non-variant queries.
Task 1: extracting the scope of the question
LLMs are very good at natural language discussions. We can essentially use the LLM to "query" the user's query (or question) to get the desired scope to call the API. For this task we can use a template as follows:
template_string = """
Given a user query, select the result scope to best suited for the query.
Return a JSON object formatted to look like:
{{
"scope": most appropriate scope to respond to the query, if unsure use "unknown"
}}
CONDITIONS
"scope" MUST be one of the candidate scope
names specified below OR it can be "unknown" if the query is not
well suited for any of the candidate scopes.
CANDIDATE SCOPES
individuals: user expects indivdual or people entries to be returned
biosamples: user expects biosample or samples entries to be returned
runs: user expects runs entries to be returned
analyses: user expects analyses entries to be returned
datasets: user expects datasets entries to be returned
cohorts: user expects cohorts entries to be returned
g_variants: user explicitly expects only genomic variants or variants entries to be returned
QUERY
{query}
OUTPUT JSON is:
"""
Note that the double braces {{ are used because this will be used as a python template string to replace {query} portion using the user's input. Let's see the rest of the code.
# langchain
from langchain.chat_models import AzureChatOpenAI
from langchain.prompts import ChatPromptTemplate
from langchain.output_parsers import PydanticOutputParser
# pydantic
from pydantic import BaseModel
from enum import Enum
# system
import os
llm = AzureChatOpenAI(
deployment_name="firstcontact",
model="gpt-4-32k"
)
class ScopeEnum(str, Enum):
biosamples = 'biosamples'
individuals = 'individuals'
runs = 'runs'
analyses = 'analyses'
genomic_variants = 'g_variants'
class ScopeQuery(BaseModel):
scope: ScopeEnum
user_query = "USER INPOUT" # <-- replace with user's question
scope_parser = PydanticOutputParser(pydantic_object=ScopeQuery)
prompt_template = ChatPromptTemplate.from_template(
template_string,
partial_variables={
"format_instructions": scope_parser.get_format_instructions()
}
)
query_scope = prompt_template.format_messages(query=user_query)
query_scope_response = llm.invoke(query_scope)
scope_query_result = scope_parser.parse(query_scope_response.content)
print(scope_query_result.json())
Upon completion the code should yield (1) g_variants for the first question and (2) individuals for the second question. Note that we use LangChain and Pydantic for creating the templates to call the LLMs and validation of LLM responses respectively. This ensures failures whenever the LLM provides us with an out-of-scope value that our API does not support. Now we have the scope and can switch to the next stage. Let's consider the case of a variant query for now.
Task 2: Extracting variant filters
We can use a strategy similar to scope extraction for this task. Refer to the following code.
template_string = """
Given a user query, extract the assembly, chromosome, start base and end base.
Return a JSON object formatted to look like:
{{
"assembly_id": what is the mentioned assembly name (eg: grch38, hg38, or something similar) if unsure use "unknown",
"chromosome": chromosome mentioned in the query (could be a number 1-22, X or Y), if unsure use "unknown",
"start": start base pair or position; single number or an array of two numbers, if unsure use "unknown",
"end": end base pair or position; single number or an array of two numbers, if unsure use "unknown",
}}
CONDITIONS
whenever a field is unsure; use "unknown"
Ensure all fields are valid JSON
QUERY
{query}
OUTPUT JSON is:
"""
class VariantQuery(BaseModel):
assembly_id: str = "unknown"
chromosome: str = "unknown"
start: list[int] | int | str = "unknown"
end: list[int] | int | str = "unknown"
For simplicity, only the template string and Pydantic model is shown.
Beyond natural language understanding
LLMs can offer far greater capabilities than just deciphering user queries into simple JSON objects. Let's consider the second scenario, where the user was making a phenotype related query. We can build a similar LangChain app as follows.
Task 3: Extracting phenotype filters
For this we can design a new template_string with necessary information related to a phenotypic query.
template_string = """
Does the query contain a set of conditions.
Return a JSON object with an array formatted to look like:
{{
"filters": [
{{
"term": place only one condition term here,
"scope": what might be the scope of this condition, chose from "individuals", "biosamples" and "runs"
}}
]
}}
CONDITIONS
filters: this is an array with objects having two attributes called "term" and "scope"
term: please insert only one condition here, if there are many, use multiple objects in "filters" array
QUERY
{query}
OUTPUT JSON is:
"""
user_query = "USER INPOUT" # <-- replace with user's question
class FilterQuery(BaseModel):
term: str
scope: ScopeEnum
class FiltersQuery(BaseModel):
filters: list[FilterQuery]
f_query_parser = PydanticOutputParser(pydantic_object=FiltersQuery)
prompt_template = ChatPromptTemplate.from_template(
template_string,
partial_variables={
"format_instructions": f_query_parser.get_format_instructions()
}
)
f_query = prompt_template.format_messages(query=user_query)
f_query_response = llm.invoke(f_query)
f_query_result = f_query_parser.parse(f_query_response.content)
print(f_query_result.json())
This yields a JSON object as follows.
{
"filters": [
{
"term": "renal failure",
"scope": "individuals"
},
{
"term": "liver damage",
"scope": "individuals"
},
{
"term": "samples isolated in broth medium",
"scope": "biosamples"
}
]
}
Note that the terms are successfully separated as three filters with appropriate scoping of them. However, a big challenge remains unanswered. Beacon API does not understand the human wordings for diseases. For example renal failure is not understood but rather SNOMED:42399005 is understood instead. For this we can leverage the embedding capabilities of LLMs along with vector databases. Luckily for us our Beacon implementation, sBeacon has a index which already stores a summary of clinical terms and their labels for faster querying.

Task 4: From natural language to ontology
Our sBeacon's terms index is a table similar to the following.
| term | label | scope |
|---|---|---|
| SNOMED:1234 | disease 1 | individuals |
| NCIT:1234 | condition 1 | biosamples |
| HP:1234 | phenotype 1 | individuals |
First, we must embed the labels in this index and create a new table with an additional column called embedding. This can be achieved as follows.
# only the additional are inputs noted below
# system
import os
import csv
embeddings_model = AzureOpenAIEmbeddings(
azure_deployment="firstcontact-embeddings",
model="gpt-4-32k"
)
if not os.path.isfile('embeddings.csv'):
entries = []
with open('./terms.csv') as fp:
reader = csv.reader(fp)
for row, (term, label, scope) in enumerate(reader):
if row==0:
continue
entries.append((term, label, scope))
to_embed = list(map(lambda x: x[1], entries))
embedded = embeddings_model.embed_documents(to_embed)
with open('embeddings.csv', 'w+') as fp:
writer = csv.writer(fp)
writer.writerow(['term', 'label', 'scope', 'embedding'])
for embedding, (term, label, scope) in zip(embedded, entries):
writer.writerow([term, label, scope, embedding])
The above code snippet create a new csv file using our original which was also provided in the form of a csv file. The embeddings can be efficiently stored in a vector database.
Vector databases are special class of databases that allows searching across a vector space efficiently using an appropriate distance metric (cosine, euclidian, etc). Note that, using such databases we intent to fetch most similar entries compared to exact matches that we exercise with traditional databases. It is also worth noting that most modern databases provide vector search capabilities due to exponential growth in GenAI applications.
Let's see how we can index our newly obtained embeddings in a vector database.
# only the additional are inputs noted below
# Vector DB
from docarray import BaseDoc
from docarray.typing import NdArray
from docarray.index import InMemoryExactNNIndex
# numerical methods
import numpy as np
docs = []
class Entry(BaseDoc):
term: str
label: str
scope: str
embedding: NdArray[1536]
with open('./embeddings.csv') as fp:
reader = csv.reader(fp)
for row, (term, label, scope, embedding) in enumerate(reader):
if row==0:
continue
embedding = eval(embedding)
docs.append(Entry(term=term, label=label, scope=scope, embedding=embedding))
db = InMemoryExactNNIndex[Entry]()
db.index(docs)
Note that for this specific example, we have used DocArray which has an in-memory database ideal for development and testing. Our entries in this database consists of . We can search for documents in this database as follows.
# embedding query
term = "renal failure"
query = np.array(embeddings_model.embed_documents([term]))
# find similar documents
matches, scores = db.find(query, search_field='embedding', limit=3)
print(f'{matches.label=}')
print(f'{matches.term=}')
print(f'{scores=}')
For illustration, we have used the first term: renal failure. We are asking for the top 3 results using the limit parameter. The output of this execution yields the following output.
matches.label=['Renal failure', 'Alcoholic liver damage', 'Hyperkalaemia']
matches.term=['SNOMED:42399005', 'SNOMED:41309000', 'SNOMED:14140009']
scores=array([0.91019983, 0.81845957, 0.80819009])
It is evident that we get the intended document with a higher score. However, it is worth noting that there may be similar false positive results, just because the two terms has some semantic similarity due to the nature of the LLM. Now this result can be utilised to auto-fill the user interface or provide the user with the choice to pick one or more of these.
Technical Considerations
In our experiments, we observed that the performance varies between language models. While larger models understood the context faster, smaller models required significantly more instructions to understand the intentions of the script.
For end-to-end executions, one can use Routing functionality of LangChain which also allows gracefully quitting the chain of thought when it is not reaching the expected outcomes in intermediate steps.
The scenario where phenotypic features are included together with variant constraints, it might require clever prompting to avoid the mingling of variant filters inside the phenotypic filter extraction stage. For these situations, we can ask the LLM to rewrite the user's question without the information we extracted already to improve accuracy. Furthermore, this can reduce the cost by progressively reducing the number of tokens along the chain.

Other Considerations
LLMs can have biases due to the nature of data it has been trained on, global trends during the training times or hallucinations. Thus it is crucial to take steps to avoid unintended outcomes such as offensive, incorrect behaviour or under-representation of traits causing mis-diagnosis.