
The Transformational Bioinformatics Group published new software to help fight COVID-19: It supports researchers in choosing the right viral strain for pre-clinical models and vaccine testing.
Developing vaccines, treatments and diagnostics are some of the key weapons in the fight against the COVID-19 pandemic. As announced in Nature, CSIRO has become an integral part in this global response by having established one of the first pre-clinical models for testing vaccine candidates [1]. This work was led by Prof S.S. Vasan and funded by The Coalition for Epidemic Preparedness Innovations (CEPI) and is part of the newly created Australian Centre for Disease Preparedness (ACDP), which will play a critical role in anticipating and preparing Australia for biosecurity threads going forward.
Genomics a game-changer for pre-clinical models
Underpinning the pre-clinical achievements was the work on understanding the genome of this novel virus; its blue-print, which holds instructions about its behaviour and what kind of disease it can cause. While SARS-CoV-2 (the virus causing COVID-19) has a proof-reading mechanism and hence mutates slower than other RNA viruses (e.g. flu), the functional consequences of the mutations it does accumulate are yet unknown.
This is discussed in the recently published peer-reviewed paper in Transboundary and Emerging Diseases (PDF pre-print) [2]. Here, the team summarises the issues involved in choosing the right strain to avoid establishing pre-clinical models with strains that might have mutations that are not representative of the circulating virus population.
To illustrate this, the team created a schematic that describes how a virus may evolve (i.e. acquire mutations) to adapt to its new human host after jumping from an animal reservoir (e.g. bats) to humans; potentially via an intermediate vector. While sequencing gives us a good visibility of this process, the individual viruses isolate from people (isolates) remain only random snapshots. For example, the first human SARS-CoV-2 isolate sequenced (with orange and pink mutations) may be genetically more different from the original strain that first infected humans (completely grey), compared to a strain that happened to be sequenced later (green). Hence aiming to pick a universal strain for animal models by minimising the distance to the first sequenced one might be the wrong approach.
There might also be more than one strain circulating (orange-pink-purple and orange-pink-brown), which in turn may be different from the most virulent one (orange-pink-blue). So, vaccine and diagnostic efforts might hence want to find isolates that represent the epidemiologically or clinically more relevant strains instead of a universal. Though it should be noted that the epidemiological relevance is often hard to quantify during ongoing outbreaks [3].
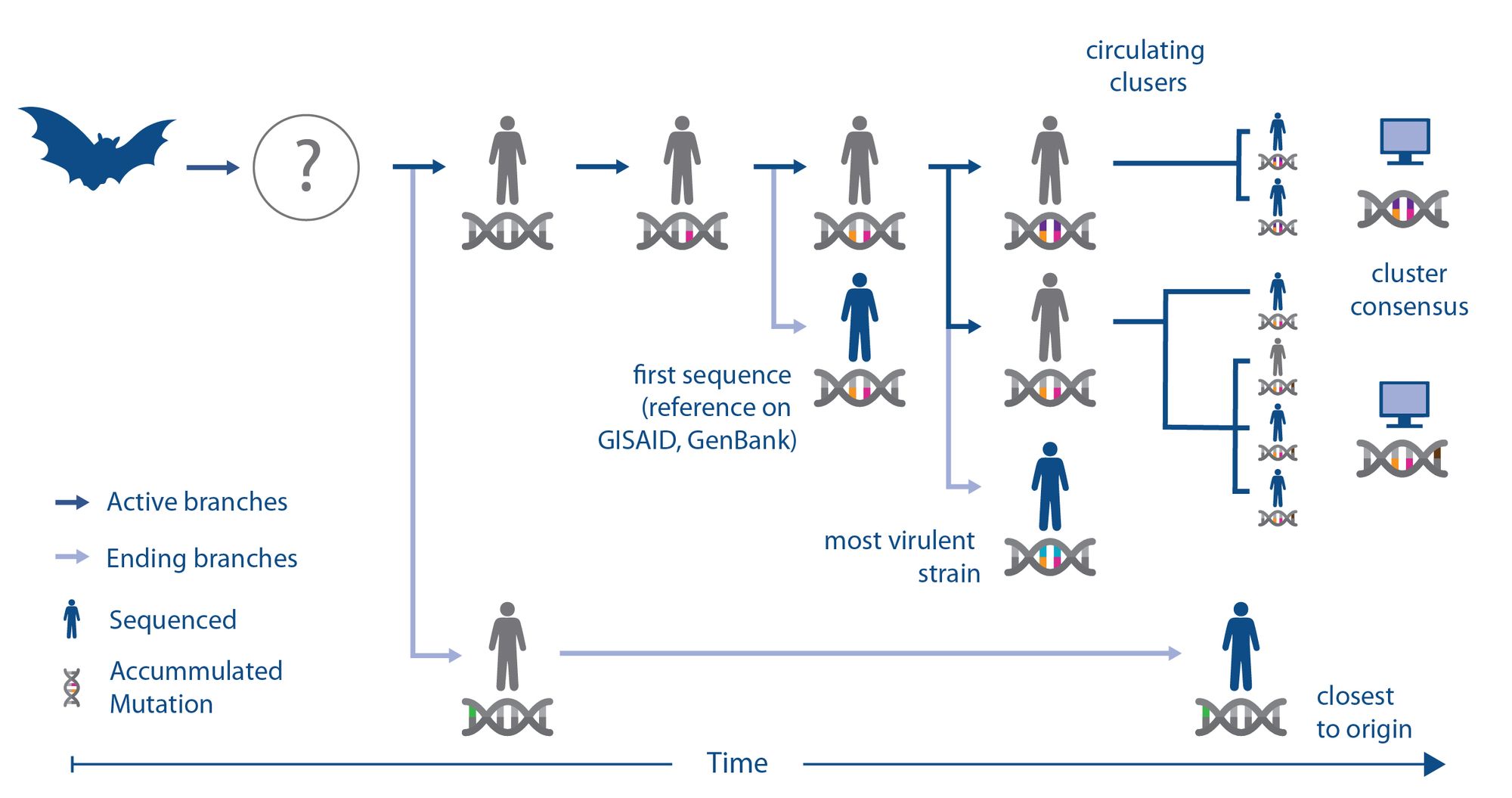
To test how much of these theoretical considerations are of concern for the current COVID-19 pandemic, the team has downloaded all viral sequences from the Global Initiative on Sharing All Influenza Data (GISAID), the repurposed de-facto repository for international SARS-CoV-2 genome sequences.
As outlined in the paper [2], from this analysis it became clear that the SARS-CoV-2 reference is likely representative of the first sequence that has infected humans, however it is also clear that there are several circulating strains with unique and stable mutations whose functional consequences are yet unknown.
Choosing representative viral strains
In the absence of clinical annotations, such as disease severity or transmissibility (which [2] calls for), choosing the right strain for pre-clinical models is a topic of active research, especially as the virus continues to evolve: choosing the reference might result in a general but out-dated model, while choosing a strain with more recently acquired mutations might result in a model that represents part of the viral population but potentially not the one with the epidemiologically or clinically relevant features.
Bauer et al. [2] argues that the right strain should have a small genetic distance to all the newly emerging isolates, hence keeping it effectiveness to a virus that has continued to evolve for the period of the clinical trials (e.g. 12-18 months). However, determining and visualising all these distances is difficult for a virus of 30,000 letters and more than 10,000 isolates.
The standard approach is to generate a phylogenetic tree to visualise the evolutionary distance. Here, sequences on different branches are assumed to be genetically more diverse than sequences on the same branch. However, phylogenic approaches are discrete, meaning if an isolate has the same number of mutations from two strains, the algorithm places it close to only one. Here, especially recombination events are problematic, where two potentially distant strains produce an offspring with merged features. Furthermore, phylogenetic trees first require a multiple-sequence alignment to be created, which for thousands of strains can quickly become computationally very expensive or impossible.
Alignment-free genomic distance measures
We have developed an alignment-free approach that can cope with the rapidly expanding viral sequence repository and scale to millions of isolates.
Here, every organism and isolate can have a unique genomic signature based on the composition of their genomic sequence. To quantify this signature, we determined the K-mer frequency. Counting of all possible strings of length k in the sequence of the virus has emerged as an alternative to phylogenetic trees in other disciplines [4]. The conceptual distance between all isolates can then be visualised by running a Principal Component Analysis (PCA) over all genomic signatures to reduce this high-dimensional K-mer frequency vector into a two-dimensional space for visualisation [5].
In the paper [2], we applied this approach to all SARS-CoV-2 sequences to map the evolutionary distance already claimed by the virus and choose a representative animal model for the Australian COVID-19 response.
Visualising the growing number of SARS-CoV-2 sequences
To enable other researchers to visualise the ever growing sequences in GISAID, we've released a web-service that offers a real-time view of the genomic landscape of the SARS-CoV-2 virus.
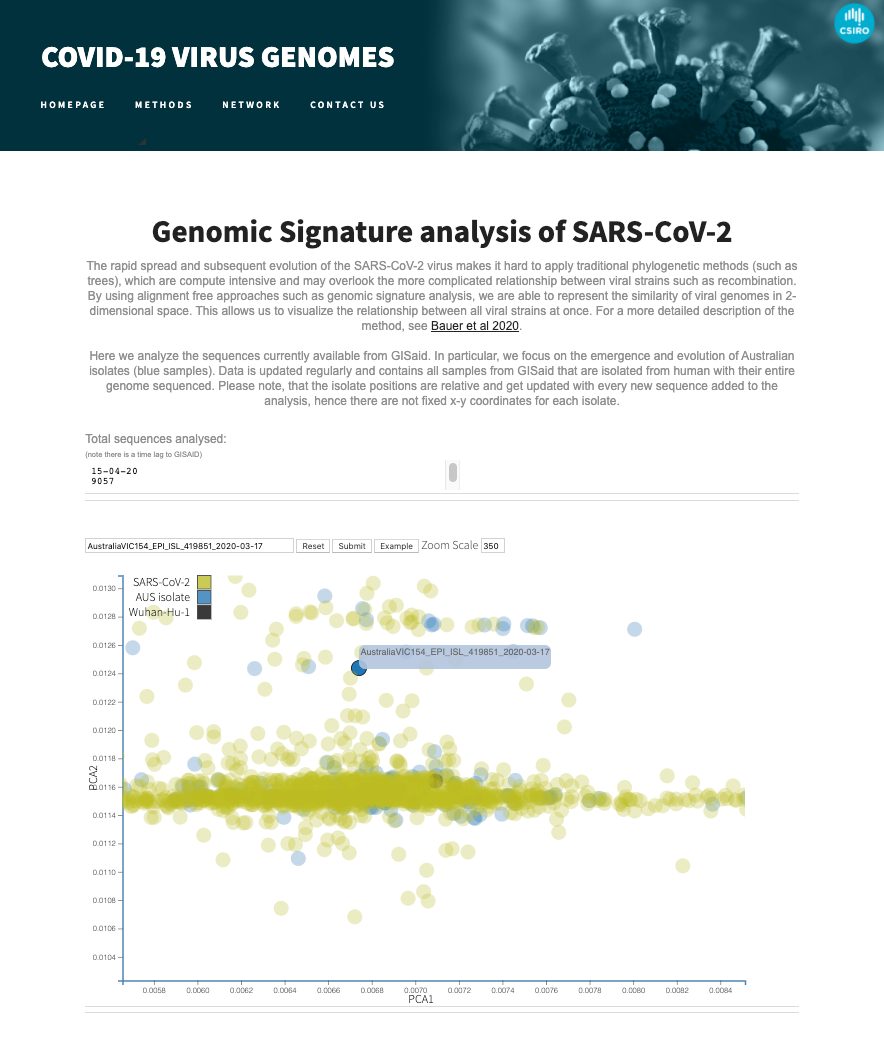
Media resources
- Transboundary and Emerging Disease paper: https://onlinelibrary.wiley.com/doi/10.1111/tbed.13588
- Paper pre-print: https://bioinformatics.csiro.au/public/files/tbed.13588.pdf
- CSIRO-press Release: https://www.csiro.au/en/News/News-releases/2020/CSIRO-unlocks-new-way-to-understand-evolving-strains-of-SARS-CoV-2
- Transformational Bioinformatics COVID-19 response overview: https://bioinformatics.csiro.au/covid19-genomics/
- Dangerous pathogens COVID-19 response overview: https://bioinformatics.csiro.au/dangerous-pathogens-team-covid-19-response/
- Australian E-health Research Centre : https://aehrc.com/covid-19/
- University of York Press Release: https://www.york.ac.uk/news-and-events/news/2020/research/evolving-strains-sars-cov-2/
- Macquarie University Press Release: https://www.mq.edu.au/newsroom/2020/04/20/macquarie-university-professor-leads-csiro-team-in-development-of-a-novel-analysis-of-covid-19-in-race-for-a-vaccine/
- Macquarie University Lighthouse Feature: https://lighthouse.mq.edu.au/article/april-2020/the-missing-piece-in-finding-a-vaccine-bioinformatics
-
Twitter
-
Linkedin
#EXCLUSIVE: About half the COVID-19 cases in Australia studied by the CSIRO have been found to be a mutant strain of the virus that may be more contagious.https://t.co/uHDc7vZJlm
— The Australian (@australian) May 8, 2020
The same coronavirus strain turned up in an Australian and a tiger. How? https://t.co/P0CBAhi9kw
— The Sydney Morning Herald (@smh) June 25, 2020
Novel Bioinformatics software for COVID-19 vaccine testing - The Transformational Bioinformatics Group published a new software tool to help fight COVID19: It supports researchers in choosing the right viral strain for pre-clinical models and vaccine ...https://t.co/WmVv0s0fAe
— TransfBioinf (@TBioinf) April 19, 2020
CSIRO identifies six COVID-19 clusters. @ssvasan and @uniOfYorkhttps://t.co/H9SW06UBEC
— Sue Dunlevy (@Sue_Dunlevy) April 19, 2020
New genetics research from Macquarie University Associate Professor Denis Bauer @allPowerde and her colleagues could help pinpoint the ideal strain of the SARS-CoV-2 virus for vaccine development: https://t.co/cxZCkUL6iq #LighthouseNewsflash #coronavirus pic.twitter.com/geOsjVrjZl
— Macquarie University (@Macquarie_Uni) April 20, 2020
Subscribe to Transformational Bioinformatics
Stay up-to-date with our progress