The genome of the SARS-CoV-2 virus is a key information source for monitoring how the virus mutates to inform vaccine development, tracking ongoing outbreaks to stay ahead of the pandemic, and diagnosing patient outcome and vaccine efficacy to keep the health system effective. This page summaries our actives in public health decision making.
Monitoring: Visualising genomic virus signatures
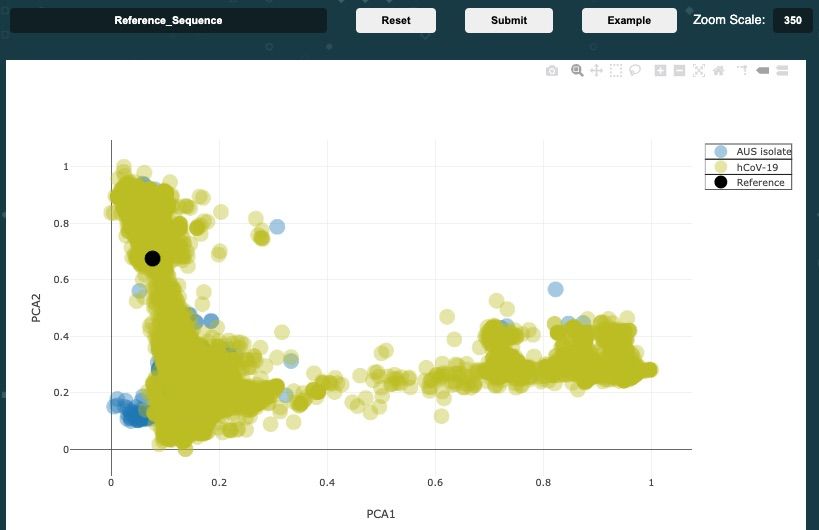
Developing an effective vaccine for evolving viruses requires us to forecast what changes the virus will have evolved by the time the vaccine is ready for role out. Taking the guesswork out of this process, we have developed a new bioinformatics approach for choosing the right viruses strain to design the vaccine against. As outlined in our Transboundary and Emerging Diseases Journal paper [1], we applied this process in the development of the first version of the COVID-19 vaccine by supporting the Australian Centre for Disease Preparedness in choosing the most representative strain to test the vaccine efficacy against. We combined large volumes of internationally available virus genomes and machine learning with laboratory observations and epidemiological insights to map the genetic diversity of the virus to visualise its evolutionary trajectory. Unlike traditional phylogenetic tools, we generate the genomic "fingerprint" of each isolate and compare it against all other samples before collapsing this high-dimensional information on a 2D map for visualization.
Based on this work, we have build a freely available visualisation page for tracking the genomic signature and their distances between virus isolates. A GitHub issue tracker is maintained and monitored to allow community volunteers to contribute.
We have since generalised the approach into the INSIDER tool [2]. Which is able to identify "foreign" pieces of genomic sequences by establishing the "normal" genetic profile for an organism to then detect stretches of sequence that does not belong. This can be applied between genomes, as done for COVID-19 viruses, or within an organism to identify antimicrobial resistance genes that were acquired through horizontal gene transfer.
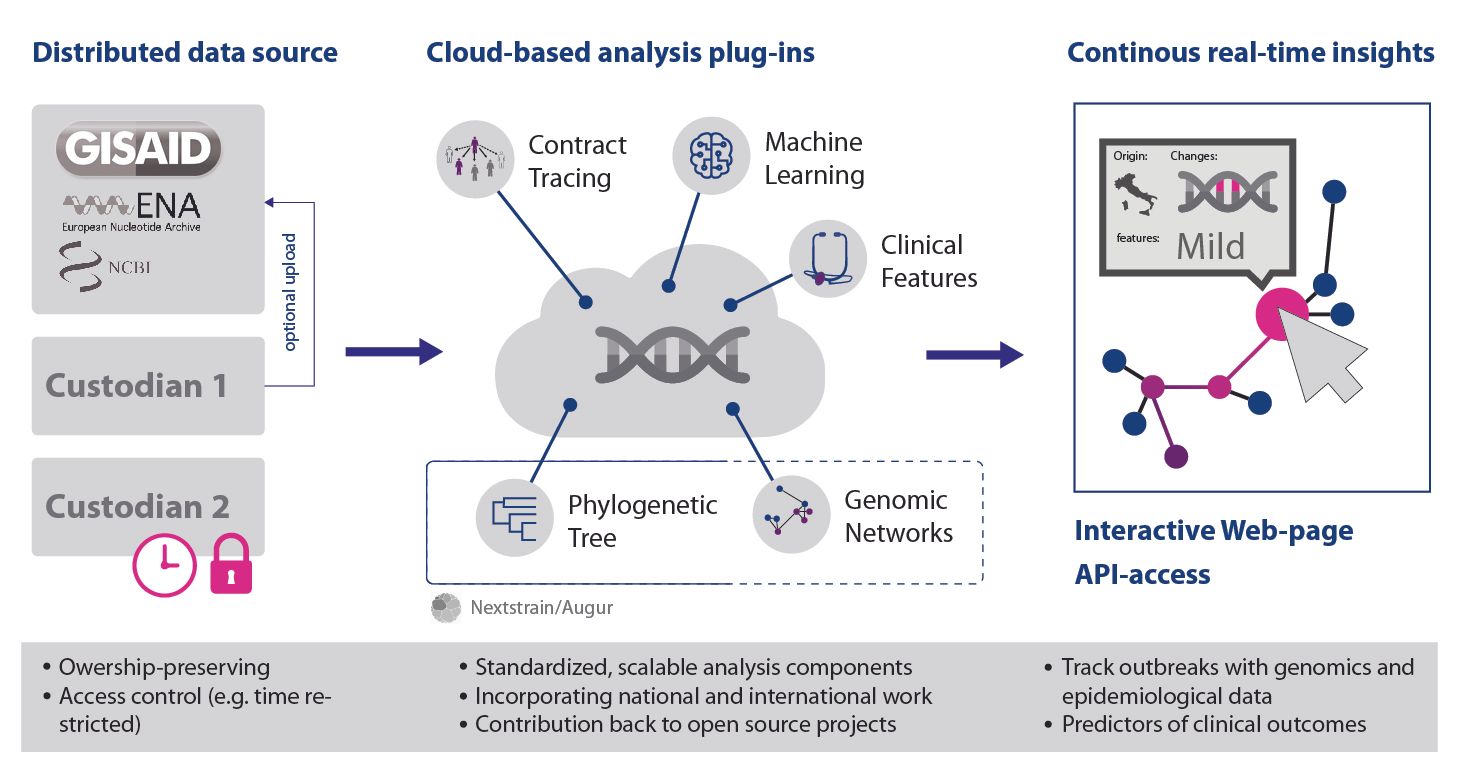
Tracking: Robust sharing and continuously analysing genomic data
Being able to track and compare emerging viral strains with historic and international records is vital to stay ahead of pandemics. Covid Beacon is based on our serverless Beacon (sBeacon) work and it enables the genomic information of a virus cohort to be queries without having to give up ownership or access control of the contributed data itself. This enables tracking of the geographical spread of a pathogenic strain, or uncovering the likely origin of an emerging strain.
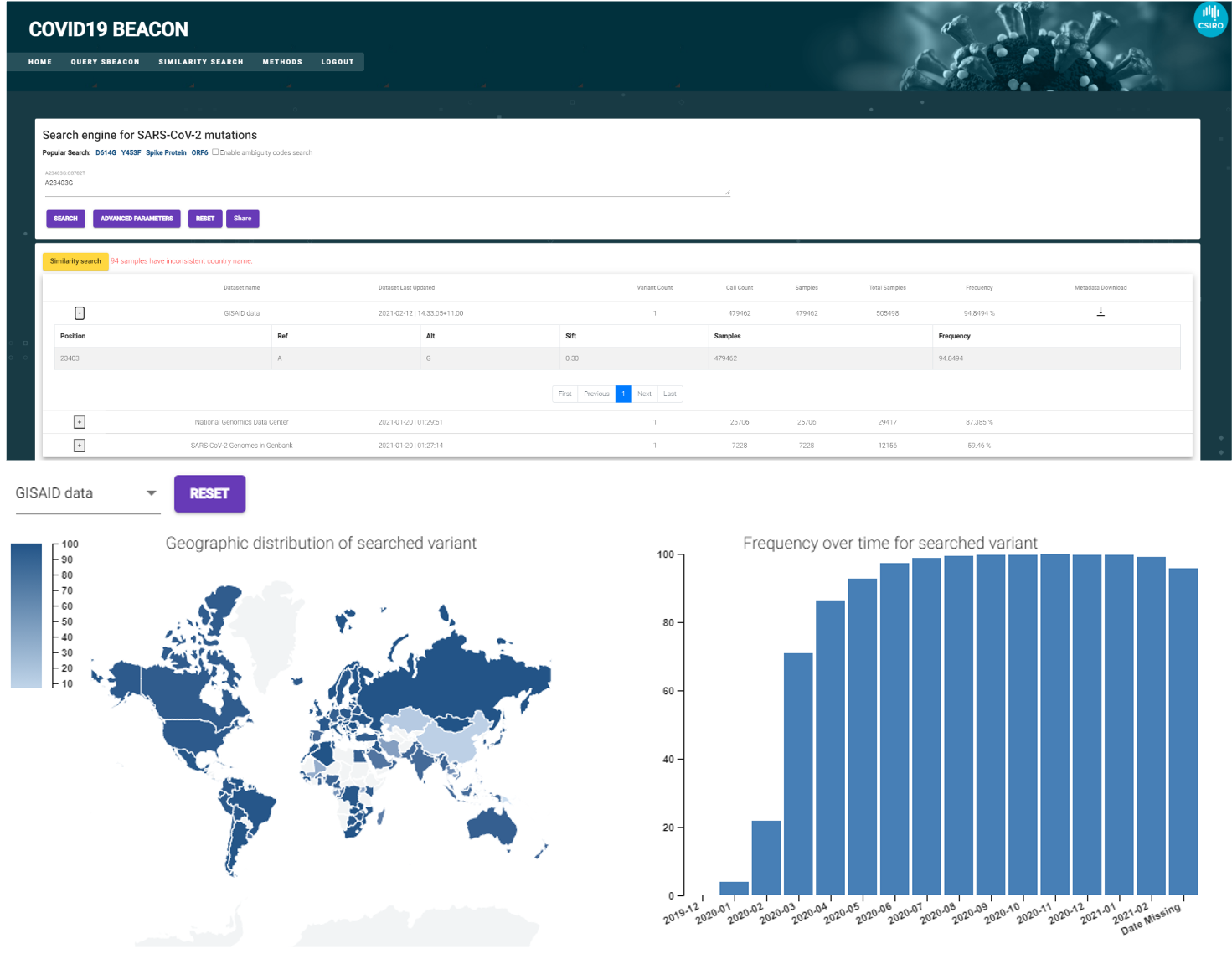
The cloud-native architecture allows the economical scaling to potentially millions of data-points and provides an appropriate environment for highly sensitive clinical data. A GitHub issue tracker is maintained and monitored to allow community volunteers to contribute.
Diagnostics: Predicting the disease outcome
COVID-19 has a wide spectrum of disease outcomes, ranging from mild or no symptoms to severe outcomes like death or long-term impairment (LongCOVID). It is currently not fully understood what the molecular determinant is of this outcome and how much the virus itself plays a role in this.
We conducted a study analyse the genomes of 10,000 COVID-19 viral samples with associated health outcomes, and found several genomic locations that are predictive of whether a patient had only a mild version of the illness or faced severe outcomes [3].
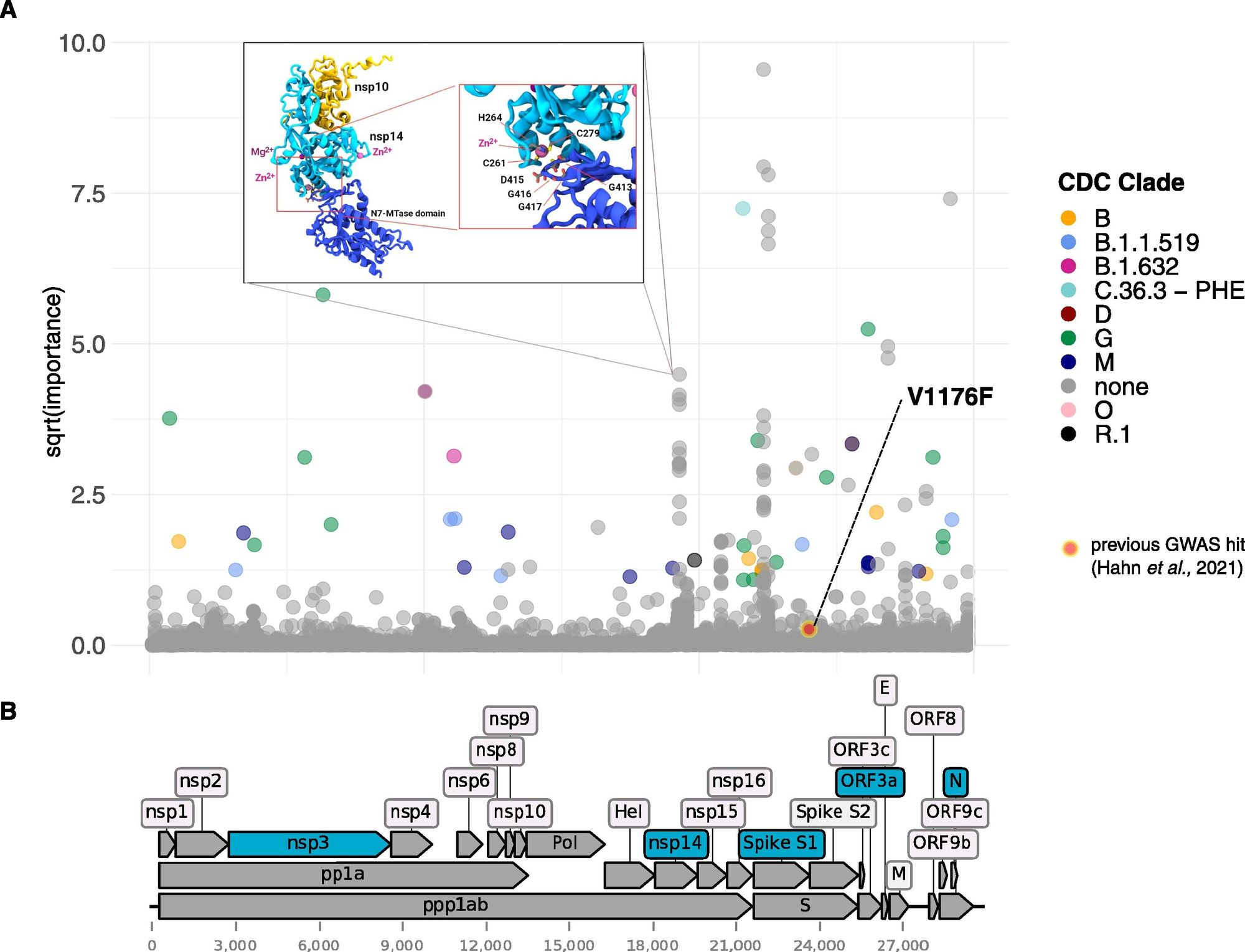
This highlights the need for capturing machine readable patient outcomes for each genomic sample captured [4]. Not only can we monitor the pathogenicity of the virus over time it will also help with quantifying the effectiveness of vaccines.
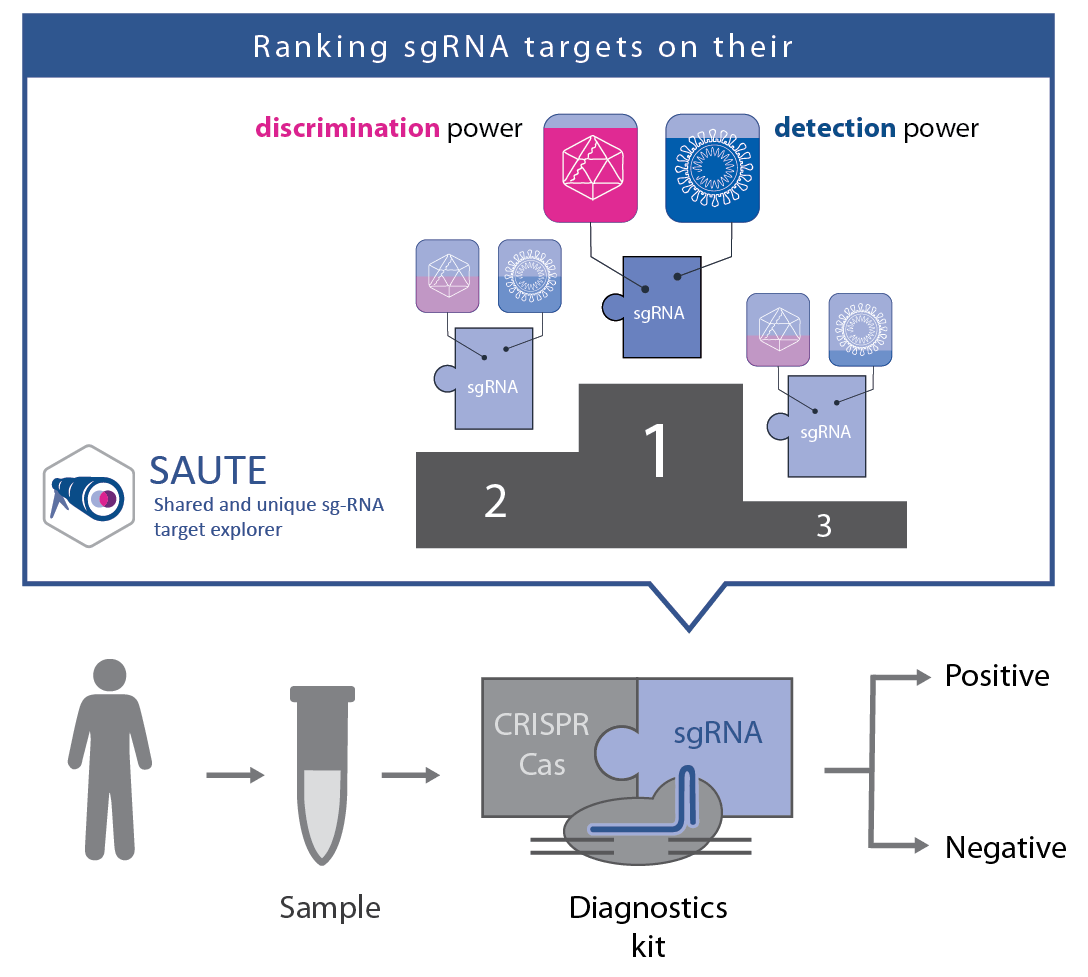
Depending on whether the virus continues to evolve into a mild and severe version there might be the need of developing strain specific at-home tests. Based on our CRISPR target-site detection tools, we built a webpage for designing CRISPR-targets that are able to differentiate between similar viruses that would form false negatives and combine variations that should be flagged as positives.