There is more genetic sequencing data than ever before, making it necessary to create approaches that enable data sharing between organzation in an efficient and secure manner. In order to address this need, the Global Alliance for Genomics and Health (GA4GH) have developed the Beacon Protocol. This protocol specifies the application programming interface (API) and the schemas used to define the entities and access framework for exchanging genomic data.
While the Beacon protocol effectively addresses challenges in data interoperability, its applications are primarily focused on human-centric data and their metadata. This necessitates adaptations during the implementation phase, especially when developing beacons for other research areas, such as pathogen genomics.
In this article, we discuss how we have addressed specific challenges in developing our version of a pathogen beacon, named PathSBeacon (Pathogen Serverless Beacon). Furthermore, we explore additional attributes that could significantly enhance the Beacon protocol's utility for researchers involved in the exchange of pathogen data.
PathSBeacon motivation
Urgency for genomic data exchange reached its most recent peak during the COVID-19 pandemic, where it was used to monitor the mutations rapidly acquired by the SARS-CoV-2 virus. While individual mutations may not necessarily have had a massive impact on the virus or the host, monitoring the variations across regions over time played a huge role in studying the evolution of the virus and the pandemic at large. This monitoring was especially useful in identifying new outbreaks and understanding the virus's migration patterns.
The urgency for such monitoring led to our development of PathSBeacon, a pathogen-focused serverless implementation of the Beacon protocol. PathSBeacon significantly differed from Beacon Protocol version 1, which prevailed at the time, due to its unique integration of metadata specific to the pathogen domain
Metadata for pathogen monitoring
In contrast to human genomics, pathogen genomics and monitoring have different assessment criteria. For example, the location of a discovery plays a more significant role compared to the geographic location of a person, which may be influenced by many social factors. Therefore, PathSBeacon uses the following metadata fields to render visualisations and statistics on pathogen data.
Sample collection date - date that the sample was taken from a patient
Location - geographical location of the sample's origin
State - location state or province (narrower geographic location)
Location + Sample collection date - combined location and date
State + Sample collection date - combined state and date
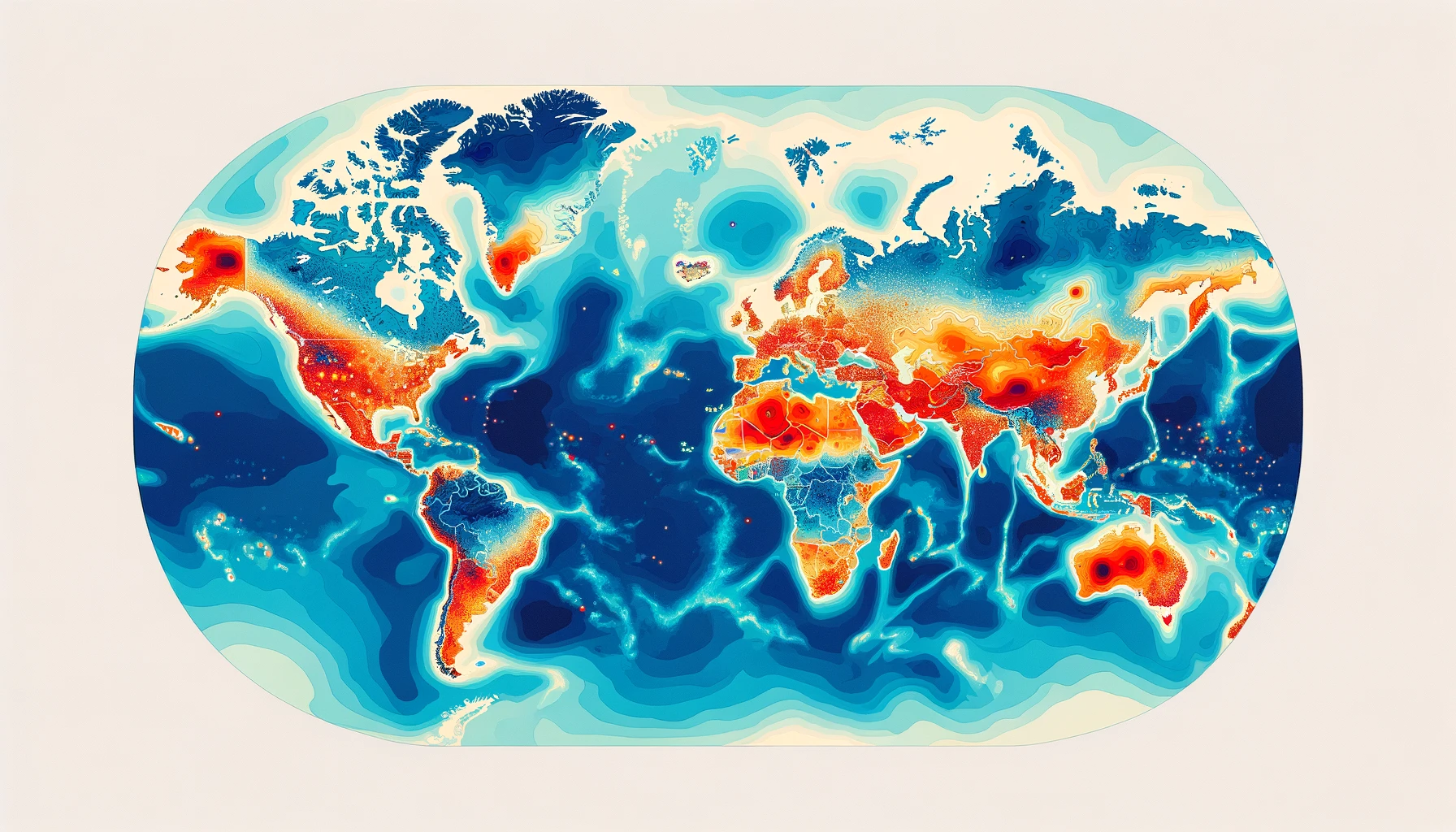
Note that the last two fields facilitate time series studies examining samples under various geographical constraints, derived from the first three fields. All values are reported along with the total counts (e.g., 10 samples include the variants of interest out of 100 total collections in Australia) to enable heatmap studies, as illustrated in the mock figure above. While these metadata entries significantly enhance analytics, we believe more improvements are possible.
Additional functionality
The manner and scale of the data over which our PathSBeacon is constructed means that we needed to make some changes to the interface and the back end. These problems included:
- Virus strains are identified by multiple mutations, rather than just a single locus. This means that we needed to make composite queries possible, where users can specify a profile of mutations, and only samples that match every mutation in the profile are returned. In addition, we support arbitrary combinations of mutations, including grouping, ANDs, ORs, and NOTs.
- Need to quickly respond to queries over upwards of 10 million genomes. The backend is able to instantly scale to thousands of worker nodes to handle multiple or larger queries, with each worker computing a small part of the total request. In addition, caching is implemented between every level of the workflow. This means if a particular mutation is present multiple times in a query, or in multiple queries, its statistics only need to be computed once. Similarly, if a profile is requested by multiple users, the samples for that profile only need to be calculated once.
- Source data can be imprecise, including ambiguity characters rather than just A, C, G, or T. We added an option to interpret IUPAC ambiguity characters in the profile or source data, allowing them to match any of their component bases. This means that a search for A23403N will yield results for any mutation at the 23403rd base, and an R (representing either A or G) will match a W (representing A or T).
- Searched profiles may be novel, or include only very few samples. We added a similarity search function that suggests subprofiles, consisting of subcombinations of mutations present in the queried profile, that have more representation in the source data. These can then be searched at the click of a button, yielding information about possible precursor strains or accounting for sequencing errors.
- Information on samples is required at different levels of granularity. While sometimes knowledge that a mutation profile is present in a location is enough, often more specific aggregation information is required. To this end, in addition to the presence or absence of a mutation profile, we also specify the number of times that profile has appeared with each metadata value, as well as the number of times that metadata value appears in the full dataset, for frequency analysis. If more specific sample-level information is needed, this can also be optionally returned. An example of this is returning the Accession ID for every sample that contains the queried profile.
Further extending the Beacon protocol for pathogen datasets
With the arrival of Beacon V2, we now have the capability to natively store metadata using the standard Beacon protocol. However, since the core of the protocol is primarily aimed at human genomics, we believe the following additions would significantly enhance the potential applications of a beacon.
Additional entities and modifications to allow generalisation
Beacon V2 currently uses two collection types: Datasets and Cohorts. It also has 5 models for storing Individual, Biosample, Run, Analysis and Genomic Variant information. We believe the following additions would be valuable in the pathogen domain.
- Host - in a pathogen beacon the centre of focus is the pathogen, therefore a host should be defined to characterise a pathogen (cow, pig, human, rice)
- Species - with an increase in scope to cover multiple species, a field to represent this is required.
- Biosamples - while biosample is defined, the model could be generalised to accommodate attributes such as geographical location (e.g., country, regions, jurisdictions), the nature of the site (tree, animal, human tissue), climate, etc.
Additional attributes in the Beacon response
It is equally important to accommodate more information in the generated response to allow direct analytical inferences.
- Frequency statistics - frequency of a given trait (genomic variant, ontology term, custom filter) in datasets or in the beacon as a whole.
- Result locations - similar to what we have already implemented in PathSBeacon
Additional querying capabilities
In order to yield the maximum potential for the new attributes and models, the protocol will also require extra querying capabilities. A few of these could be as follows.
- Filter by frequency - one might be interested in searching for pathogens that show more than 50% abundance across samples from hosts with a certain disease.
- Search within a geographic region - while ontologies can facilitate this at the country/state level, being able to search around an outbreak location given a distance might make more sense in certain applications. These can include creating fine-grained heatmaps or contour plots to detect epicentres or monitor the spread of an outbreak.
Although a single instance of a beacon can concurrently handle human and pathogen data, it is advisable to segregate them into different beacon instances. While this may seem like it would increase compute resource costs, our serverless approaches, such as sBeacon and PathSBeacon, have demonstrated otherwise. Organisations can deploy as many beacons as they wish (COVID-sBeacon, TB-sBeacon, etc.) without incurring any additional costs.
Find out more on our product page: https://bioinformatics.csiro.au/pathsbeacon/